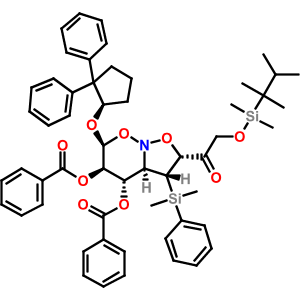
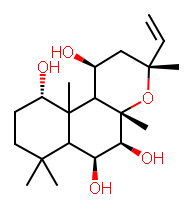
Wouldn't it be nice to be able to copy a molecule from a PDF into a molecular drawing package? Well, here are some instructions for doing this on Windows with BKChem, and using OSRA to do the conversion. Click on the image to the right for a screenshot of this in action.
Wouldn't it be nice to be able to copy a molecule from a PDF into a molecular drawing package? Well, here are some instructions for doing this on Windows with BKChem, and using OSRA to do the conversion. Click on the image to the right for a screenshot of this in action.(1) Install Python 2.6 (or just use 2.4 or 2.5 if you have one of these already)
(2) Install the Python Imaging Library 1.1.6 for your version of Python
(3) Download and extract BKChem-0.12.5.zip
(4) Drop convert_clipboard_image.py and convert_clipboard_image.xml into the BKChem plugins folder (Note: if the webserver is down, you can get these files here and here)
(5) Download and extract osra-mingw-1-1-0.zip
(6) Set the environment variable OSRA to the full path to osra.exe
(7) Find the Snapshot tool (it has a picture of a camera) in your version of Adobe Reader. In version 9 it's under Tools/Select and Zoom/Snapshot Tool, and you can add it to the toolbar under Views/Toolbars/More Tools.
(8) Open a PDF of a paper containing a molecular structure (e.g. Figure 4 in this paper of mine), and use the Snapshot tool to draw a box around a molecule and hit CTRL+C to copy (if not done automatically).
(9) Start BKChem by double-clicking on bkchem.py (in the bkchem subfolder)
(10) Click "Plugins", "Paste and Convert Image"
Notes:
(0) Open Source software allows you to implement crazy ideas as fast as you can think of them.
(1) This won't work with the latest exe release of BKChem as py2exe didn't include the ImageGrab module (part of PIL).
(2) For this to work with ChemDraw, I need to know how to place ChemDraw XML (which I can create with OpenBabel) on the clipboard so that ChemDraw will be able to paste it. (To be clear, I know how to place it on the clipboard in general, it's just how to place it in such a way that ChemDraw will recognise it as a chemical structure. Hmmm...just found this...)
(3) Bond angles are perturbed slightly (e.g. vertical bonds can become skewed). Maybe this can be fixed on the OSRA side.
(4) The hamburger reference and some background can be found on PMR's blog.
(5) Thanks to Leonard (see comment below) for the information on the Snapshot tool in Adobe Reader.